Research Interests
The research interests of our group include the total synthesis of biologically active natural products, the development of new synthetic methods, heterocyclic chemistry, bioorganic and medicinal chemistry, combinatorial chemistry, the study of DNA-agent interactions, and the chemistry of antitumor antibiotics. We place a special emphasis on investigations to define the structure-function relationships of natural or designed agents in efforts to understand the origin of their biological properties.
As the exploration of the properties of complex natural products becomes increasingly more sophisticated with the technological advances being made in their screening and evaluation and as structural details of their interaction with biological targets becomes more accessible, the importance and opportunities for providing unique solutions to complex biological problems has grown. A powerful complement to the examination of the naturally-derived agents themselves is the preparation and subsequent examination of key partial structures, agents containing deep-seated structural modifications, and the corresponding unnatural enantiomers of the natural products. Well conceived deep-seated structural modifications may be used to address the structural basis of the natural products interactions with biological targets and to define fundamental relationships between structure, functional reactivity, and properties. In these studies we address the challenging problem of understanding the beautiful solutions and subtle design elements that nature has provided in the form of a natural product and work to extend the solution through rational design elements to provide more selective, more efficacious, or more potent agents designed specifically for the problem or target under investigation.
Central to such studies is the development of dependable synthetic strategies and the advent of new synthetic methodology to permit the preparation of the natural products, key partial structures, and analogs incorporating deep-seated structural changes. The resulting efforts have reduced many difficult or intractable synthetic challenges to manageable problems providing an approach not only to the natural product but one capable of simple extrapolation to a series of structural analogs as well. In our own efforts this has provided the opportunity to fully explore the origin of the natural products properties and to devise agents with improved selectivity and efficacy.
Our ongoing investigations emphasize the development and application of hetero Diels-Alder reactions, the thermal reactions of cyclopropenone ketals, inter- and intramolecular acyl radical-alkene addition reactions, medium and large ring cyclization procedures, and the benzannulation reaction of arylchromium carbene complexes. In each instance, the methodology development represents the investigation of chemistry projected as a key step in the total synthesis of a natural or nonnatural product. Most recognized of these efforts include the inverse electron demand Diels-Alder reactions of heterocyclic azadienes (1,2,4,5-tetrazines, 1,2,4-triazines, 1,2,3,5-tetrazines, 1,3,5-triazines, 1,2,3-triazines, 1,2-diazines, including the first reported (1982) organocatalytic Diels-Alder reaction), the introduction of the first general method for catalyzing heterocyclic azadiene [4+2] cycloaddition reactions (solvent H-bonding of HFIP, 2016), the sucessful introduction of 1,2,3-triazine cycloaddition reactions (2011), and the first reported synthesis and cycloaddition reactions of 1,2,3,5-tetrazines (2019) that are widely used today not only in organic synthesis but also in bioorthogonal conjugation reactions. We developed the inverse electron demand Diels-Alder reactions of acyclic 1-azadienes (N-sulfonyl-1-azabutadienes, identified transition state anomeric effect), discovered the cascade [4+2]/[3+2] cycloaddition reactions of 1,3,4-oxadiazoles, explored the cycloaddition reactions of cyclopropenone ketals ([4+2] cycloaddition) and discovered of their reversible thermal (80 oC) ring opening to π-delocalized singlet vinylcarbenes and subsequent cycloaddition reaactions ([1+2], [3+2], and [4+3] cycloadditions). We also introduced acyl radical generation from phenylselenoesters and their use in subsequent intermolecular and intramolecular alkene addition reactions, discovered and developed key palladium-catalyzed reactions including the first examples of a Pd(0)-mediated free amine amination of an aryl halide (1984), unsymmetrical 2,2'-bipyrrole coupling via electrophilic Pd(II) C-H activation (1987), and intramolecular Pd(0)-catalyzed indole (macro)cyclization (2009), provided key contributions to the free radical hydrogen atom transfer (HAT) functionalization, cyclization, or reduction of unactivated alkenes (2009), and we introduced the concept of divergent total synthesis (1984).
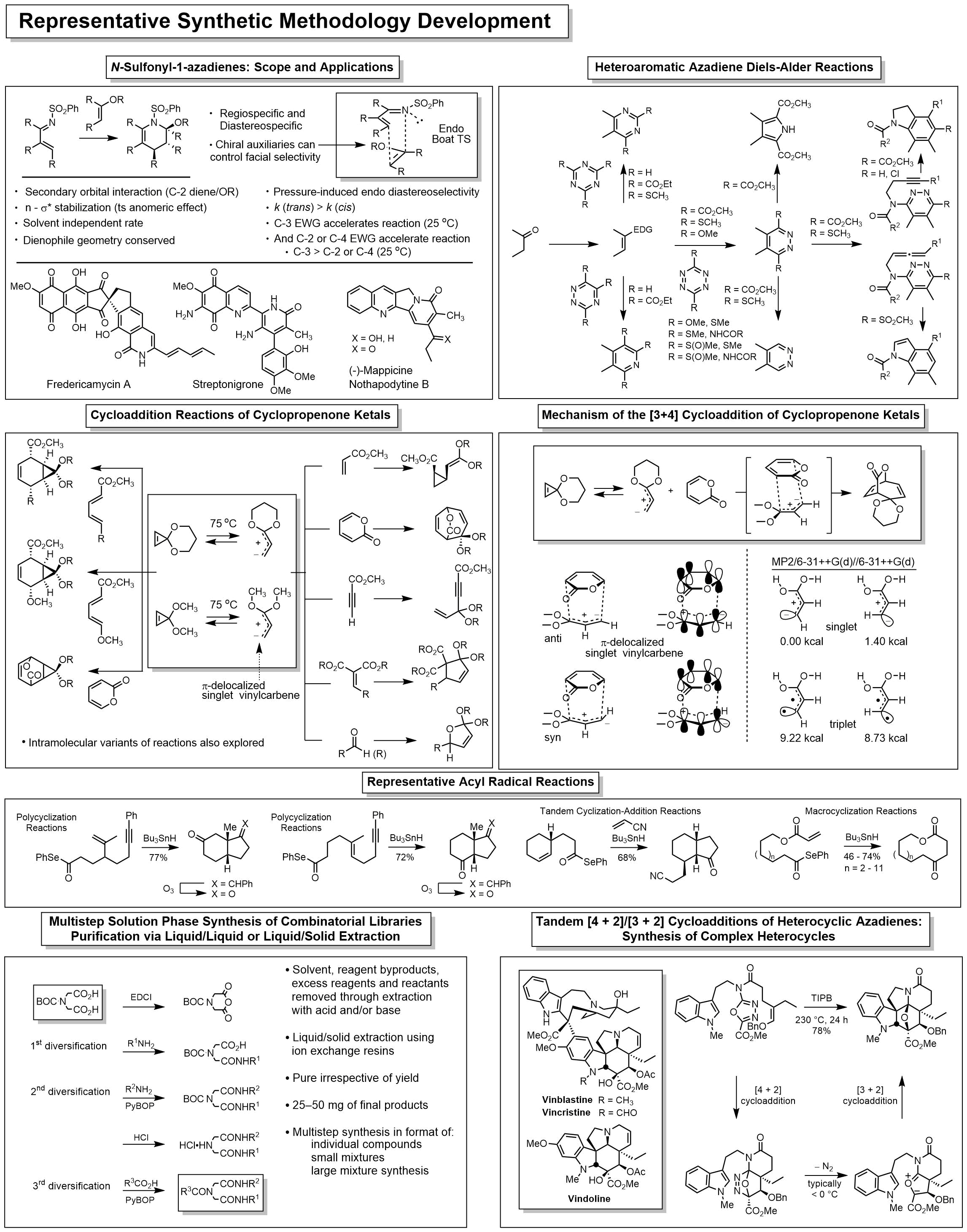
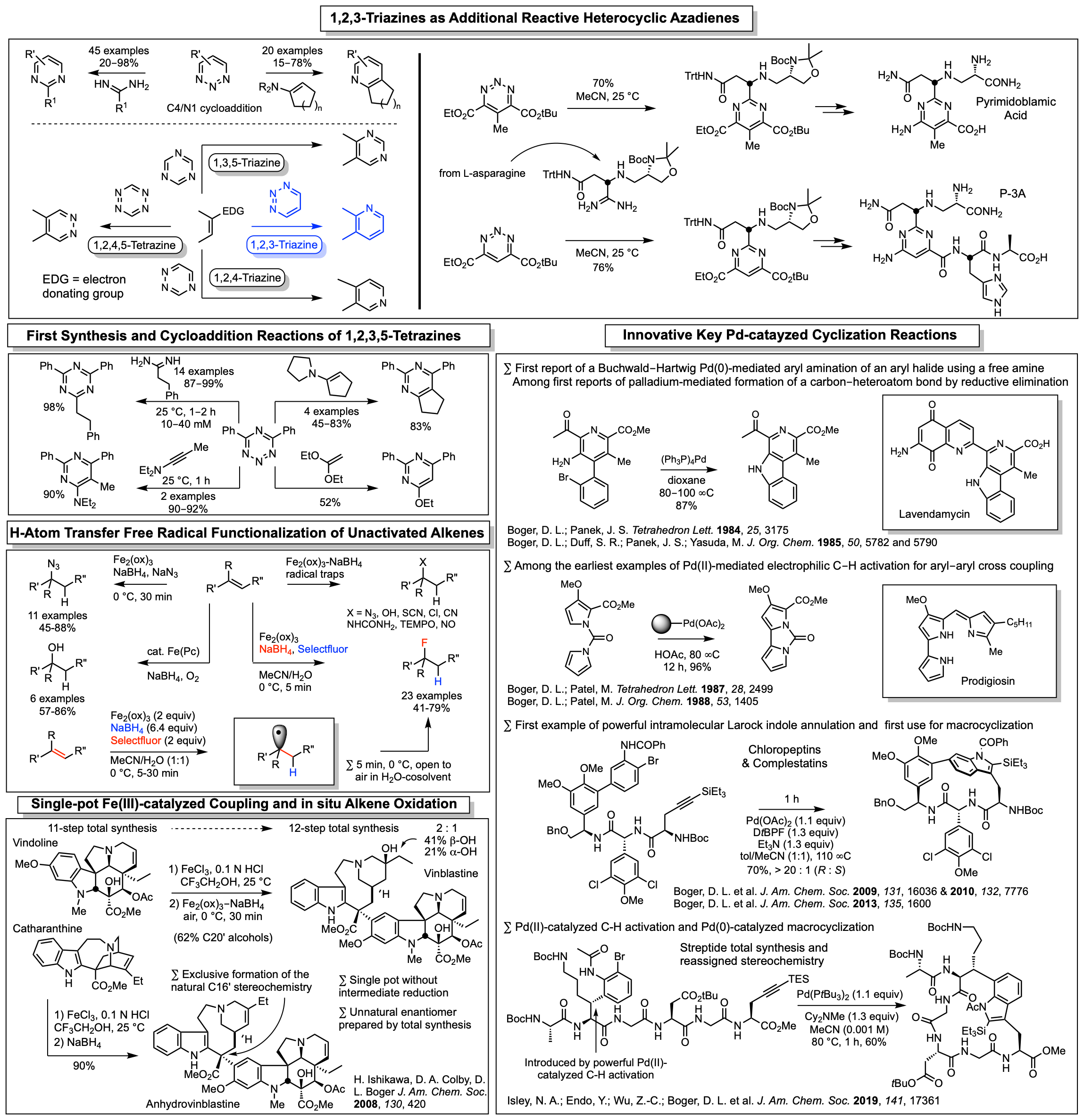
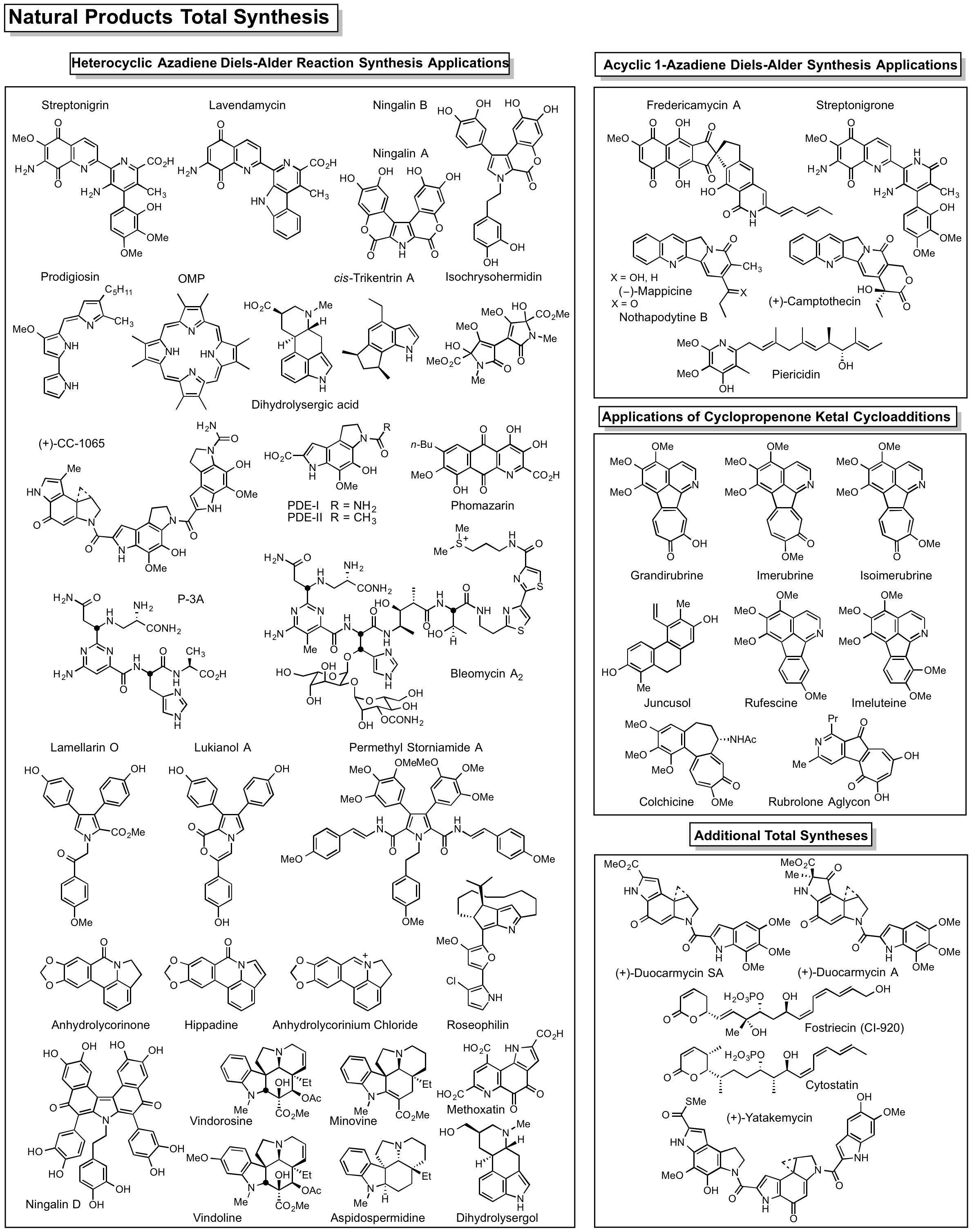
Natural Products Total Synthesis
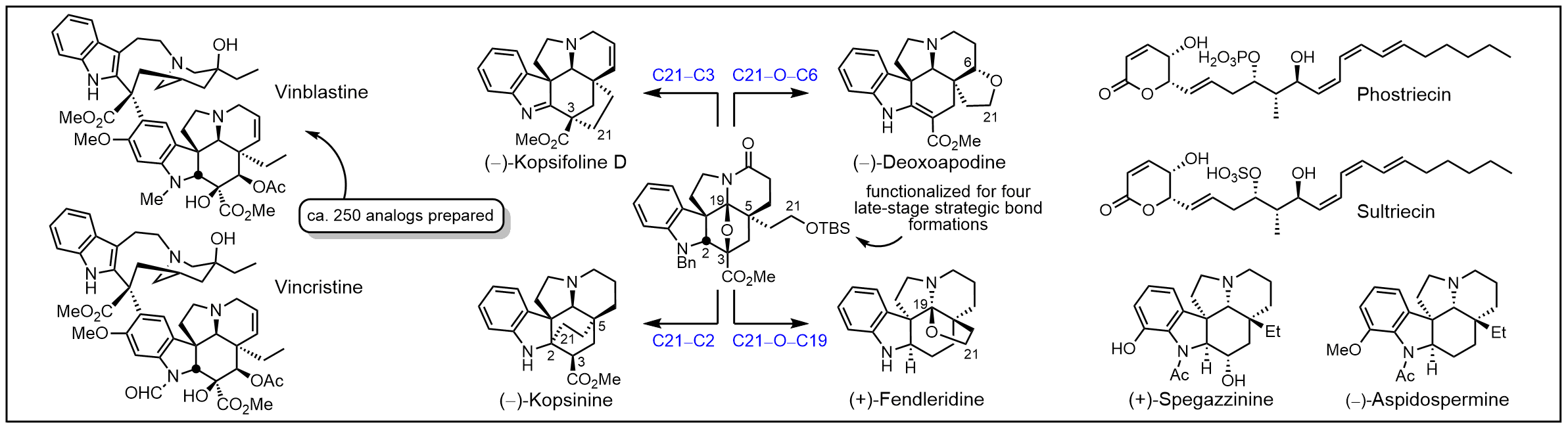
We have pursued a longstanding program targeting the total synthesis of complex, biologically active natural products chosen by virtue of their properties, and the development of new synthetic methodology or synthetic strategies designed for the natural products of interest. With this technology in hand, the studies are extended to the total synthesis of key analogs bearing deep-seated structural changes used to define the structure-function properties of the natural product, to identify its biological target if unknown, and to define the fundamental features of its interaction with its biological target. Many are first total syntheses, defining or correcting the stereochemistry or structure, and often they constitute creative, concise, efficient total syntheses easily identifiable with our efforts. Highlights include the total syntheses of steptonigrin (1983), juncusol (1984), rufescine and imelutine (1984), colchicine (1985), lavendamycin (1985), PDE-I and PDE-II (1987), (+)-CC-1065 (1988), prodigiosin and prodigiosene (1988), cis-and trans-trikentrin A (1991), combretastatin D2 (1991), (+)-duocarmycin SA (1992), (-)-pyrimidoblamicc acid (1993), streptonigrone (1993), isochrysohermidin (1993), (+)-P-3A (1994), bleomycin A2 (1994), fredericamycin (1995), grandirubrine and imerubrine (1995), (+)-duorcarmycin A (1996), nothapodytine B and (-)-mappicine (1998), ningalin A, lamellarin O, lukianol A, storniamide A (1999), phomazarin (1999), ningalin B (2000), distamycin and library of 2600 analogs (2000), hippadine (2000), rubrulone (2000), fostriecin (2001), (-)-roseophilin (2001), (+)-camptothecin (2002), anhydrolycorinone (2002), (+)-yatakemycin (2004), second generation asymmetric synthesis of (+)-yatakemycin and (+)-duocarmycin SA (2006), minovine (2005), piericidin A1 and B1 (2005), ningalin D (2005), (-)-vindorosine (2006), (-)-vindoline (2006), N-methylaspidospermidine (2006), cytostatin (2006), (+)-vinblastine (2008), (+)-vincristine (2009), phostriecin (2010), (+)-fendleridine and (+)-acetylaspidoalbidine (2010), asymmetric synthesis of (-)-vindoline and (-)-vindorosine (2010), lycogarubin C and lycogalic acid (2010), (-)-aspidospermine (2012), (+)-spegazzinine (2012), kopsinine (2013), second generation asymmetric synthesis of (+)-P-3A and (-)-pyrimidoblamic acid (2014), (-)-kopsifoline D (2014), (-)-deoxoapodine (2014), (-)-kopsinine (2015), dihydrolysergol and dihydrolysergic acid (2015); second generation synthesis of (-)-vindoline (2015); methoxatin (2016).
Natural Products Total Synthesis References
Chemical & Engineering News article on Ramoplanin (May 13, 2002)
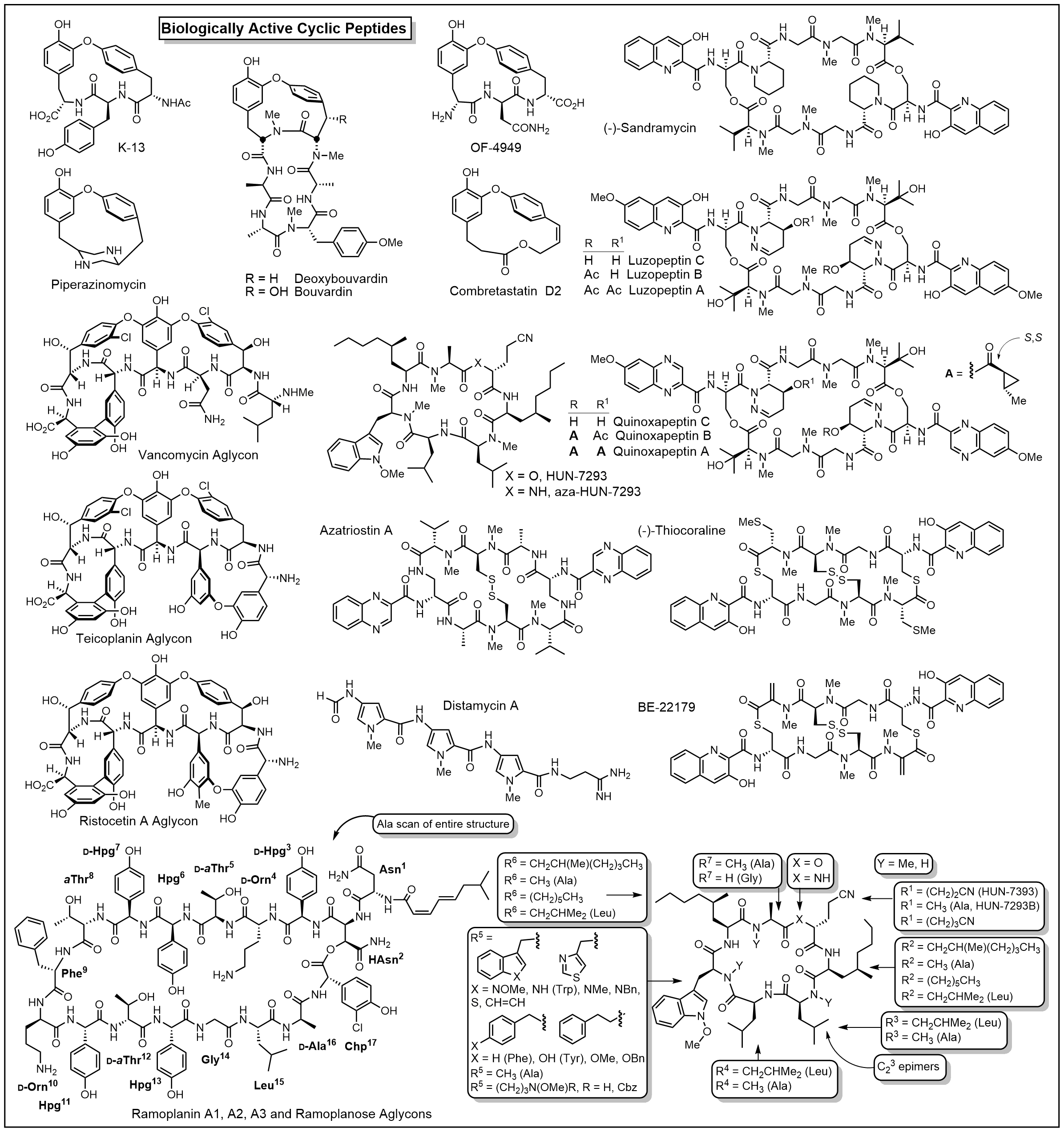
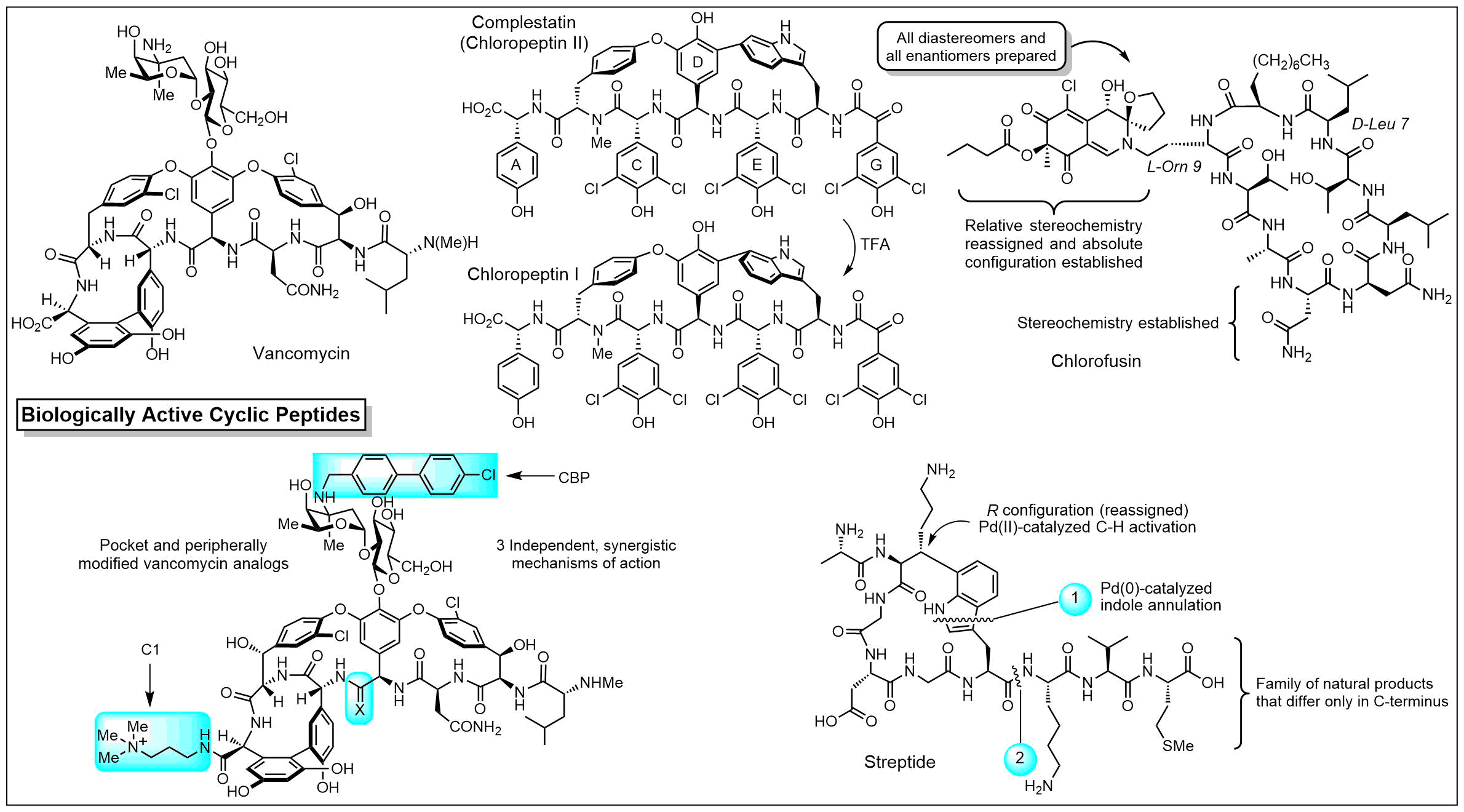
Biologically Active Cyclic Peptides
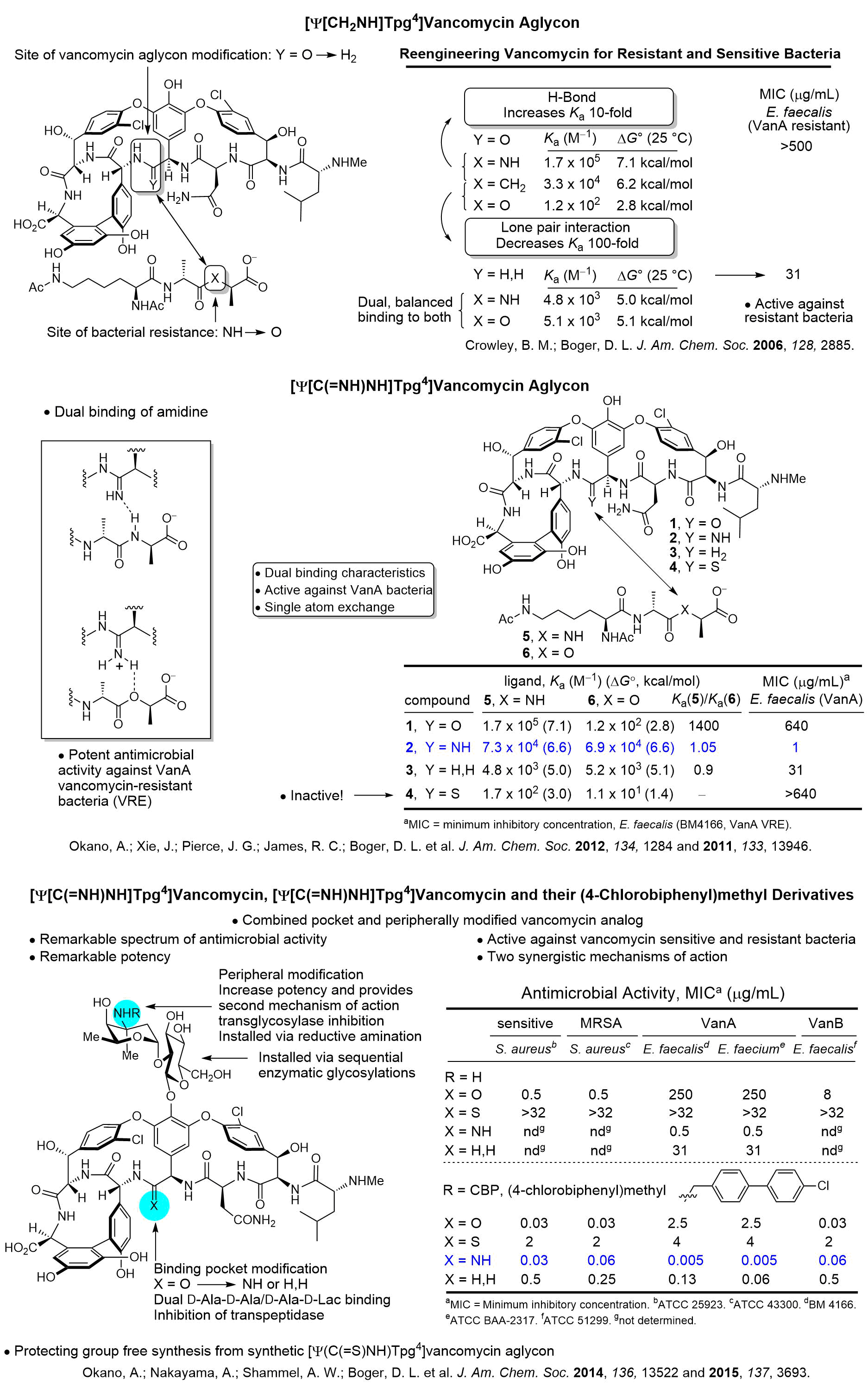
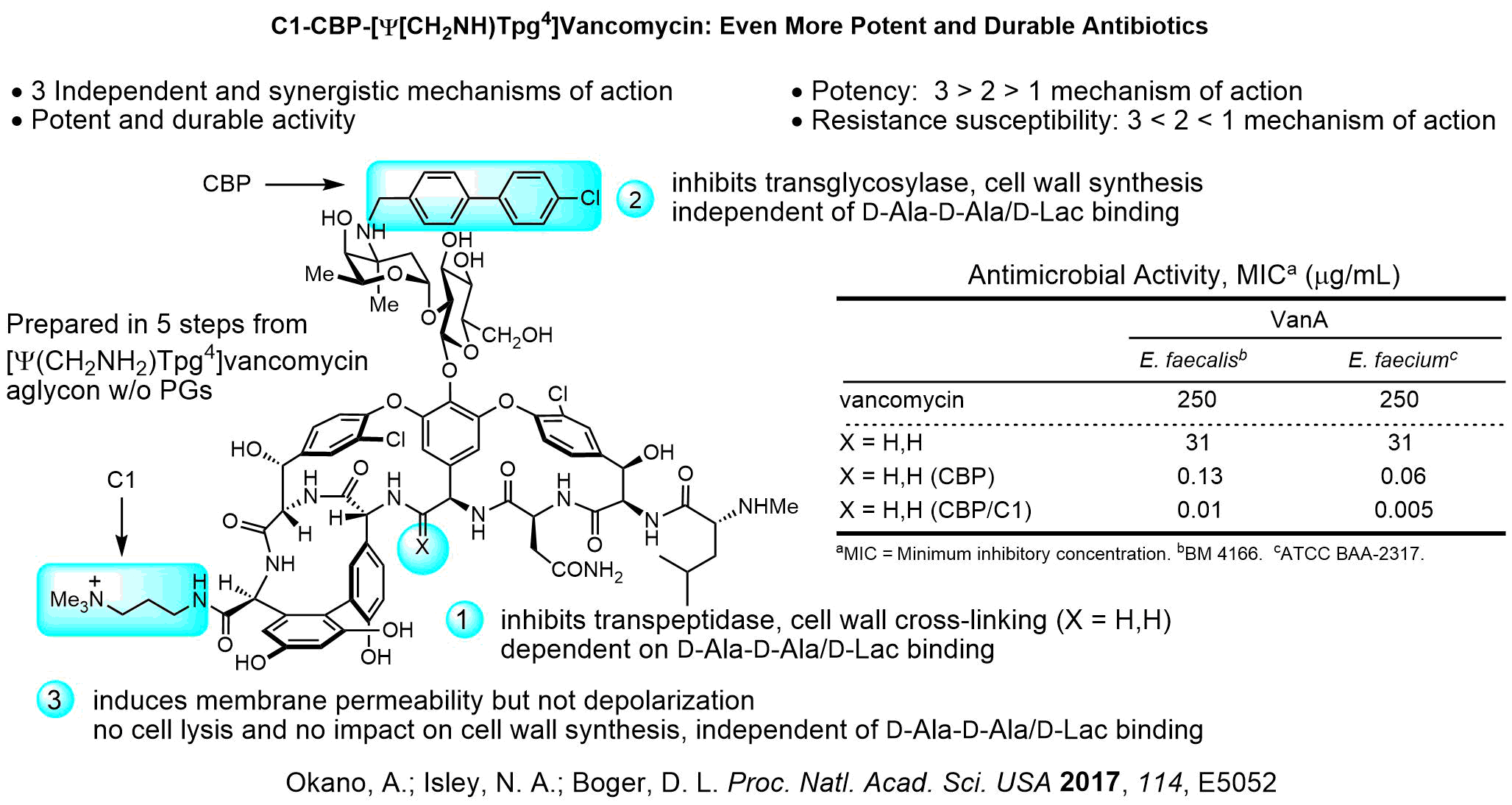
We have a long standing program on the total synthesis and evalutaion of naturally occuring biologically active cyclic peptides. This has included efforts culminating in the total syntheses of a series of antitumor agents that act as DNA bis-intercalating drugs including sandramycin (1996), luzopeptins A-C (1999), quinoxapeptins A-C (1999), thiocoraline (2000), BE-22179 (2000), and triostin A (2000), inhibitors of protein synthesis including cycloisodityrosine (1997), deoxybouvardin (1991), bouvardin (1993), and RA-VII (1993), inhibitors of p53/MDM2 binding including chlorofusin (2007) and its seven unnatural diastereomers (2008), as well as the natural products K-13, OF4949 III and IV (1990), piperazinomycin (1993), and HUN-7293 (1999), including its Ala scan library of analogs (2002). The relative and absolute stereochemistry of several of these 20 natural products were established by the total syntheses and subsequent seminal studies and their structure-activity relationships were conducted. However, the most recognized of our efforts in this area are the total syntheses of the glycopeptide antibiotics including vancomycin (2014) and its aglycon (1999), teicoplanin aglycon (2000), ristocetin A aglycon (2005), ramoplanin A1-A3 aglycons (2003) and a 17-membered Ala scan library of analogs (2007), chloropeptins I and II (complestatin, 2009), complestatins A & B (2011), and streptide (2019). In these latter studies, we also introduced and utilized a powerful intramolecular Pd(0)-catalyzed indole annulation for macrocyclization of complex cyclic peptides (2009). Recently, these studies have focused on the rational redesign of vancomycin to achieve dual binding to both D-Ala-D-Ala and D-Ala-D-Lac, altering a single atom in the binding pocket, such that the new analogs are active against both vancomycin-sensitive (e.g., MRSA) and vancomycin-resistant bacteria (e.g., VRSA, VRE). In these studies, we defined the origin of the destabilized binding to the altered D-Ala-D-Lac target in resistant bacteria (100-fold derived from repulsive lone pair/lone pair interaction, 10-fold from lost H-bond; 2003). This led to the design and total synthesis of [Ψ[CH2NH]Tpg4]vancomycin (2015) and [Ψ[C(=NH)NH]Tpg4]vancomycin (2014) and earlier their aglycons (2006, 2011). The recent total syntheses of their corresponding chlorobiphenyl derivatives have provided glycopeptide antiobiotic analogs: (1) that contain synergistic binding pocket and peripheral modifications, (2) that are endowed with two or three independent mechanisms of action only one of which is dependent upon D-Ala-D-Ala binding, (3) that display a broad spectrum of activity (e.g.; MRSA, VanA and VanB, VRE) at remarkable potencies (MICs = 0.01-0.005 μg/mL), and (4) that are especially durable antibiotics capable of extensive use in the clinic for years (2015).
Glycopeptide Antibiotics
Press Articles:
[Ψ(CH2NH)Tpg4]Vancomycin Aglycon
Science Editorial: (Volume 311, pg 321)
Article from Nature (Volume 439, pg 766)
Article from Chemical and Engineering News (Volume 84, Number 7, pg 15)
Article from Nature Reviews: Drug Discovery (Volume 5, pg 281)
Article from Microbe Magazine (Volume 1, pg 219)
[Ψ(C(=NH)NH)Tpg4]Vancomycin Aglycon
Science Editorial: (Volume 333, pg 1202)
Article from Chemical and Engineering News (Volume 89, Number 34, pg 9)
ACS Chemical Biology Spotlight (2011, 6, pg 873)
Nature Chemical Biology Highlight (2011, 7, pg 654)
[Ψ(C(=NH)NH)Tpg4]Vancomycin and its (4-Chlorobiphenyl)methyl derivative
http://www.scripps.edu/newsandviews/i_20140922/boger.html
http://cen.acs.org/articles/92/i39/Tweaked-Vancomycin-Kils-Two-Ways.html
C1-CBP-[Ψ(CH2NH)Tpg4]Vancomycin
Press Articles:
Article from San Diego Union-Tribune
DNA-Drug Interactions
A subset of investigations address DNA binding, alkylation, and cleaving agents that exhibit antitumor activity. These studies include work on CC-1065, duocarmycin A and SA, and yatakemycin where we not only conducted total syntheses of the natural products, defining the absolute stereochemistry and correcting a misassigned structure (yatakemycin), but also characterized their DNA alkylation properties. In these studies, we defined their DNA alkylation selectivity (including that their unnatural enantiomers), rates, reversibility, stereoelectronically-controlled reaction regioselectivity, isolated their adenine N3 adducts, defined the source of their DNA alkylation selectivity (noncovalent AT-rich binding selectivity - shape selective recognition), identified the source of catalysis for the DNA alkylation reaction (DNA binding induced conformation change disrupting the stabilizing vinylogous amide conjugation - shape dependent catalysis), provided high-resolution NMR-derived structures of the natural products bound to DNA, and established that they are subject to an exquisite "target-based activation". More than 135 publications and more than 2000 analogs of the natural products (e.g., CBI) have been disclosed containing deep-seated structural changes used to define relationships between structure and reactivity or structure and activity, and their contributions to the expression of the DNA alkylation properties and biological activity of the natural products (e.g. "hydrophobic binding-driven-bonding" and the predictive parabolic relationship between reactivity and the biological potency). Analogous studies on bleomycin (30 publications, ca. 100 analogs probing each substiutent and each subunit in structure) confirmed the origin of DNA cleavage selectivity derived from G triplex-like H-bonding in the minor groove, defined fundamental conformational properties contributing to the efficiency of DNA cleavage, and provided a high resolution NMR-derived structure of DNA bound deglycobleomycin A2. We prepared a >9000-membered screening library of distamycin analogs, discovered and defined the DNA cross-linking properties of ischrysohermidin and established the origin of its selectivity, and have studied the naturally occuring bis-intercalators (sandramycin, luzopeptins, quinoxapeptins, thiocoraline, BE-22179), defining their DNA binding selectivity, its origins, kinetics of binding, and established a high resolution NMR-derived structure of sandramycin bound to DNA. In the course of these studies, we introduced the powerful fluorescent intercalator displacement (FID) assay for establishing DNA binding selectvity or affinity that complements footprinting and a convenient M13-derived alternative to 32P-end-labeling of restriction fragments for DNA cleavage studies.
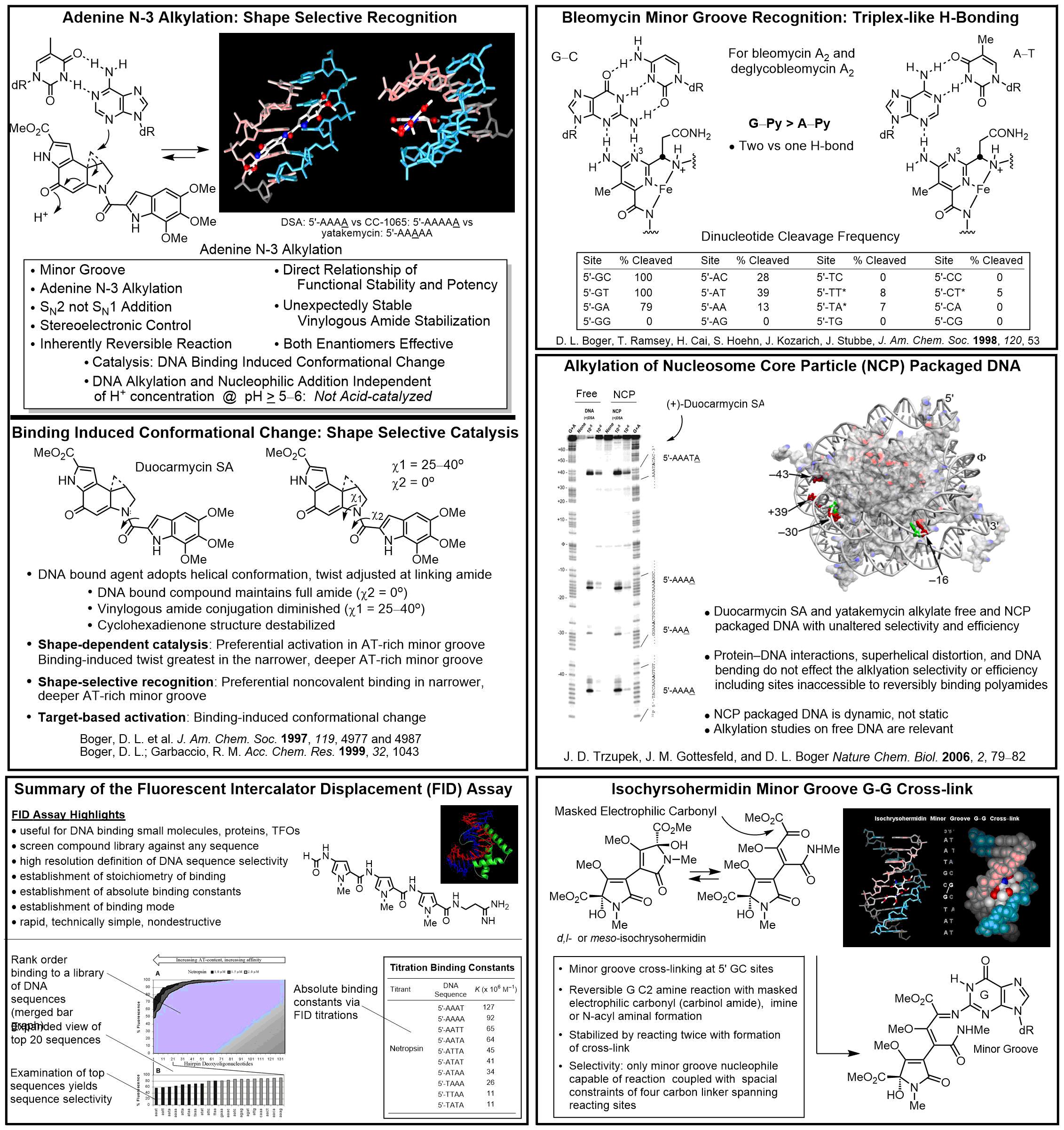
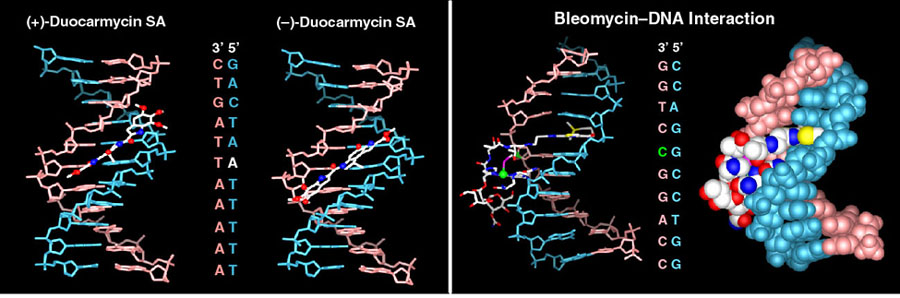
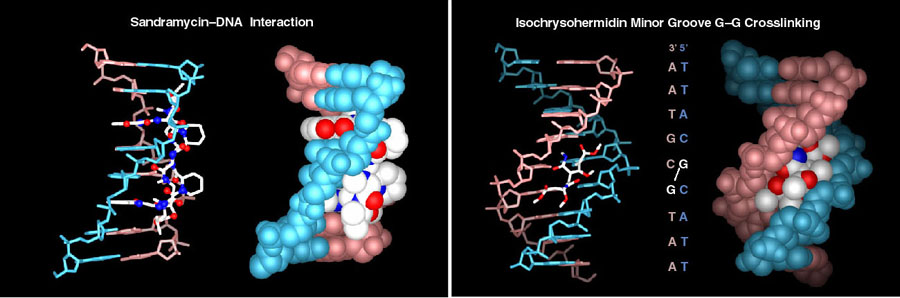
Story in Modern Drug Discovery (September 2001)
Oleamide: An Endogenous Sleep-inducing Lipid
We have been involved in a long standing program that emerged from the discovery of oleamide as an endogenous signaling molecule (1995) promoting physiological sleep, representing the first of a growing class of endogenous signaling fatty acid primary amides (46 publications to date). This led to the discovery and subsequent characterization of the enzymes responsible for their release (PAM) and degradation (e.g., fatty acid amide hydrolase, FAAH, 1996), development and optimization of orally active, long acting, potent, selective and reversible alpha-ketoheterocycle inhibitors of serine hydrolases including FAAH, introduction of a powerful proteome-wide ABPP-based selectivity screen for reversible enzyme inhibitors (2003), characterization of inhibitor bound FAAH X-ray structures, and in vivo validation of FAAH as a candidate therapeutic target. This work showed that preventing the enzymatic hydrolysis of an endocannabinoid (anandamide, endogenous signaling molecule) provides an effective and especially attractive approach for the therapeutic intervention for the treatment of pain that avoids the side effects of a blunt force agonist acting on the target receptors (CB1 and CB2). Since this only potentiates an activated signaling pathway by increasing the concentration and duration of action of the released signaling molecules at its site of action, it provides a temporal and spatial pharmacological control not available to a receptor agonist. It is the work that has inspired subsequent efforts to target enzymes controlling endocannabinoid signaling for the treatment of pain and inflamation. These studies are conducted with complementary, uniquely successful, long time collaborations at Scripps (Cravatt, Lerner) or elsewhere (Lichtman).
San Diego Union-Tribune Article on Oleamide Research at Scripps
Article from Discovery Channel Canada

Signal Transduction
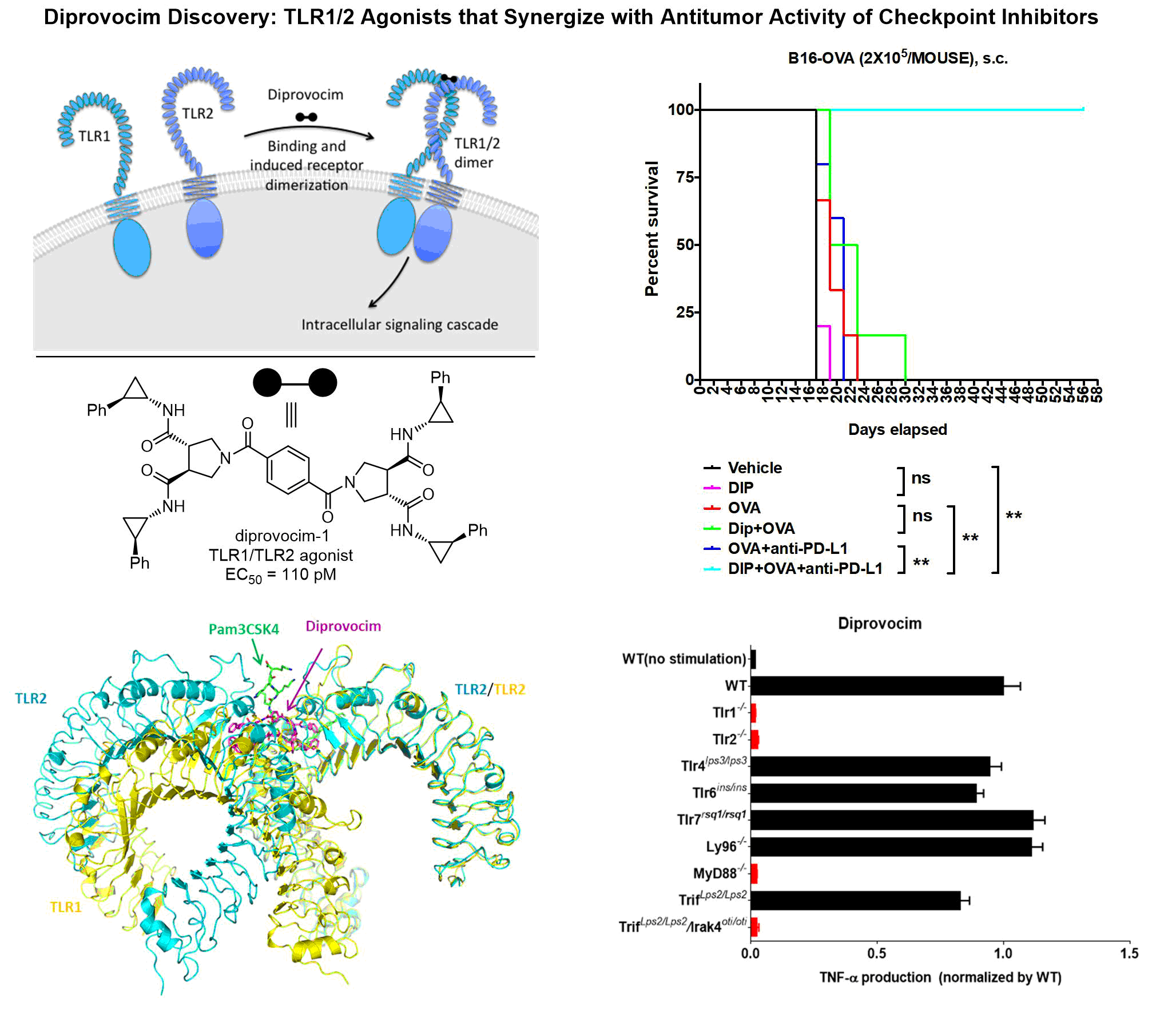
Receptor activation by homodimerization, heterodimerization, and higher order homo- and hetero-oligomerization has emerged as a general mechanism of initiating intracellular signal transduction. Studies have been initiated to investigate the fundamental principles and structural features that are embodied in such receptor activation events with the erythropoietin receptor (EPOr) and toll-like receptors (TLRs), the latter for stimulation of an immune response.
Solution-phase Combinatorial Chemistry
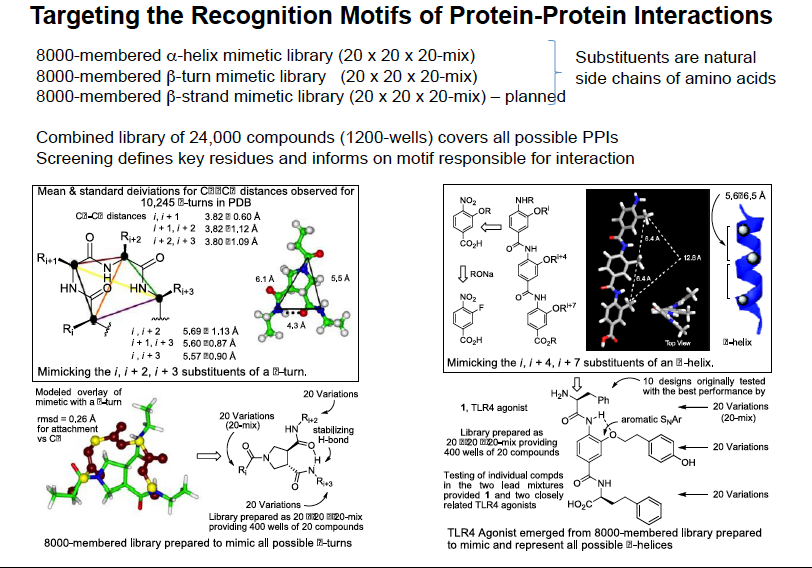
We are actively involved in the synthesis of compound libraries for target validation (40 publications) where we pioneered the use of solution-phase synthesis techniques, being the first to introduce liquid-liquid and liquid-solid (ion exchange) extraction for compound isolation and purification, using both divergent and introducing convergent synthetic strategies for library synthesis, and enlisting single compound, positional scanning, deletion synthesis and small mixture library synthesis formats. We have prepared designed libraries targeting protein-protein interactions (comprehensive 8000-membered alpha-helix mimetic and beta-turn mimetic libraries), protein-DNA interactions (9000-membered DNA binding library), and the major enzyme classes (comprehensive serine hydrolase inhibitor library). Notable applications include the first inhibition of an enzyme function by targeting its localization (inhibiting MMP2 binding and blocking angiogenesis and tumor growth in vivo, 2001), the first inhibition of a transcription factor dimerization and function by a small molecule (Myc/Max dimerization inhibiting Myc-driven cell transformation, 2002), small molecule erythropoetin (EPO) mimetics that act as agonists promoting EPO receptor dimerization and activation, 2002), enzyme inhibitors that act by inhibiting requisite enzyme dimerization (ATIC, 2005), inhibitors of aberrant gene trascription and cell transformation by blocking a protein-DNA interaction (LEF-1/beta-catenin, 2009), and have provided small molecule agonists of toll-like receptors promoting TLR4 (2016, neoseptin) and TLR1/TLR2 (2017, diprovocim) receptor dimerization and activation.
GAR and AICAR Transformylase Inhibitors
Employing X-ray crystallographic structures (I. A. Wilson, Scripps) of the apo enzymes and complexes of the enzymes with substrates (GAR, AICAR), folate cofactors, and in-house inhibitors, the novo design and examination of potential potent enzyme inhibitors as antineoplastic agents are being pursued.